Since ASU
⋯
Coming soon!
Prior Publications
-
Evaluating NaREMgWO6 (RE = La, Gd, Y) Doubly Ordered Double Perovskites as Eu3+ Phosphor Hosts
Inorganic Chemistry 2016, 55 (23), 12383-12390. DOI: 10.1021/acs.inorgchem.6b02295
Abstract
Three doubly ordered double perovskites NaREMgWO6 (RE = La, Gd, Y) have been synthesized via traditional solid-state methods, doped with Eu3+, and characterized to evaluate their promise as Eu3+ phosphor hosts. NaYMgWO6, a new member of the family, was found to crystallize in the P21 space group and is isostructural with NaREMgWO6. Emissions characteristic of Eu3+ ions (5D0 → 7F4,3,2,1,0) were observed, with the most intense transition being the 5D0 → 7F2 transition near 615 nm. Substitution of Eu3+ onto a more compressed RE site in the NaY1–xEuxMgWO6 and NaGd1–xEuxMgWO6 hosts results in a blue shift of the charge-transfer excitation band and an increase in the intensity of the 5D0 → 7F2 transition compared to NaLa1–xEuxMgWO6. All of the hosts can incorporate high concentrations of Eu3+ before concentration quenching is observed. When the rare-earth ion is either Gd3+ or Y3+, good energetic overlap between the Eu3+ charge-transfer band and the absorption of the host lattice results in sensitization and energy transfer from the perovskite host lattice to the Eu3+ activator sites. These hosts display comparable if not better luminescence than Y2O3:Eu3+, a commonly used commercial standard, demonstrating their promise as red phosphors.

-
Quaternary Pavonites A1+xSn2-xBi5+xS10 (A+ = Li+, Na+): Site Occupancy Disorder Defines Electronic Structure
Inorganic Chemistry 2018, 57 (4), 2260-2268. DOI: 10.1021/acs.inorgchem.7b03091
Abstract
The field of mineralogy represents an area of untapped potential for the synthetic chemist, as there are numerous structure types that can be utilized to form analogues of mineral structures with useful optoelectronic properties. In this work, we describe the synthesis and characterization of two novel quaternary sulfides A1+xSn2-xBi5+xS10 (A+ = Li+, Na+). Though not natural minerals themselves, both compounds adopt the pavonite structure, which crystallizes in the C2/m space group and consists of two connected, alternating defect rock-salt slabs of varying thicknesses to create a three-dimensional lattice. Despite their commonalities in structure, their crystallography is noticeably different, as both structures have a heavy degree of site occupancy disorder that affects the actual positions of the atoms. The differences in site occupancy alter their electronic structures, with band gap values of 0.31(2) eV and 0.07(2) eV for the lithium and sodium analogues, respectively. LiSn2Bi5S10 exhibits ultralow thermal conductivity of 0.62 W m–1 K–1 at 723 K, and this result is corroborated by phonon dispersion calculations. This structure type is a promising host candidate for future thermoelectric materials investigation, as these materials have narrow band gaps and intrinsically low thermal conductivities.

-
A New Three-Dimensional Subsulfide Ir2In8S with Dirac Semimetal Behavior
Journal of the American Chemical Society 2019, 141 (48), 19130-19137. DOI: 10.1021/jacs.9b10147
Abstract
Dirac and Weyl semimetals host exotic quasiparticles with unconventional transport properties, such as high magnetoresistance and carrier mobility. Recent years have witnessed a huge number of newly predicted topological semimetals from existing databases; however, experimental verification often lags behind such predictions. Common reasons are synthetic difficulties or the stability of predicted phases. Here, we report the synthesis of the type-II Dirac semimetal Ir2In8S, an air-stable compound with a new structure type. This material has two Dirac crossings in its electronic structure along the Γ–Z direction of the Brillouin zone. We further show that Ir2In8S has a high electron carrier mobility of ∼10 000 cm2/(V s) at 1.8 K and a large, nonsaturating transverse magnetoresistance of ∼6000% at 3.34 K in a 14 T applied field. Shubnikov de-Haas oscillations reveal several small Fermi pockets and the possibility of a nontrivial Berry phase. With its facile crystal growth, novel structure type, and striking electronic structure, Ir2In8S introduces a new material system to study topological semimetals and enable advances in the field of topological materials.

-
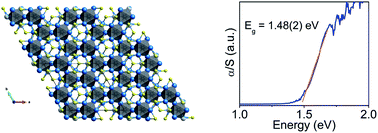
Ir6In32S21, a Polar, Metal-Rich Semiconducting Subchalcogenide
Chemical Science 2020, 11 (3), 870-878. DOI: 10.1039/C9SC05609B
Abstract
Subchalcogenides are uncommon, and their chemical bonding results from an interplay between metal–metal and metal–chalcogenide interactions. Herein, we present Ir6In32S21, a novel semiconducting subchalcogenide compound that crystallizes in a new structure type in the polar P31m space group, with unit cell parameters a = 13.9378(12) Å, c = 8.2316(8) Å, α = β = 90°, γ = 120°. The compound has a large band gap of 1.48(2) eV, and photoemission and Kelvin probe measurements corroborate this semiconducting behavior with a valence band maximum (VBM) of −4.95(5) eV, conduction band minimum of −3.47(5) eV, and a photoresponse shift of the Fermi level by ∼0.2 eV in the presence of white light. X-ray absorption spectroscopy shows absorption edges for In and Ir do not indicate clear oxidation states, suggesting that the numerous coordination environments of Ir6In32S21 make such assignments ambiguous. Electronic structure calculations confirm the semiconducting character with a nearly direct band gap, and electron localization function (ELF) analysis suggests that the origin of the gap is the result of electron transfer from the In atoms to the S 3p and Ir 5d orbitals. DFT calculations indicate that the average hole effective masses near the VBM (1.19me) are substantially smaller than the average electron masses near the CBM (2.51me), an unusual feature for most semiconductors. The crystal and electronic structure of Ir6In32S21, along with spectroscopic data, suggest that it is neither a true intermetallic nor a classical semiconductor, but somewhere in between those two extremes.
-
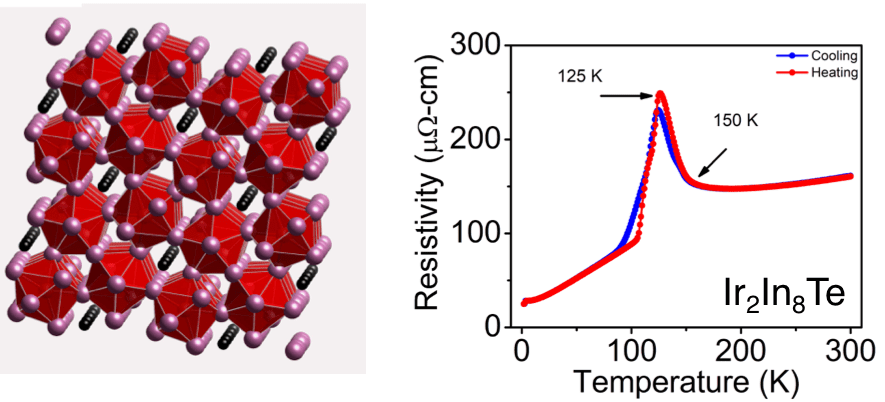
The Subchalcogenides Ir2In8Q (Q = S, Se, Te): Dirac Semimetal Candidates with Re-entrant Structural Modulation
Journal of the American Chemical Society 2020, 142 (13), 6312-6323. DOI: 10.1021/jacs.0c00809
Abstract
Subchalcogenides are uncommon compounds where the metal atoms are in unusually low formal oxidation states. They bridge the gap between intermetallics and semiconductors and can have unexpected structures and properties because of the exotic nature of their chemical bonding as they contain both metal–metal and metal–main group (e.g., halide, chalcogenide) interactions. Finding new members of this class of materials presents synthetic challenges as attempts to make them often result in phase separation into binary compounds. We overcome this difficulty by utilizing indium as a metal flux to synthesize large (millimeter scale) single crystals of novel subchalcogenide materials. Herein, we report two new compounds Ir2In8Q (Q = Se, Te) and compare their structural and electrical properties to the previously reported Ir2In8S analogue. Ir2In8Se and Ir2In8Te crystallize in the P42/mnm space group and are isostructural to Ir2In8S, but also have commensurately modulated (with q vectors q = 1/6a* + 1/6b* and q = 1/10a* + 1/10b* for Ir2In8Se and Ir2In8Te, respectively) low-temperature phase transitions, where the chalcogenide anions in the channels experience a distortion in the form of In–Q bond alternation along the ab plane. Both compounds display re-entrant structural behavior, where the supercells appear on cooling but revert to the original subcell below 100 K, suggesting competing structural and electronic interactions dictate the overall structure. Notably, these materials are topological semimetal candidates with symmetry-protected Dirac crossings near the Fermi level and exhibit high electron mobilities (∼1500 cm2 V–1 s–1 at 1.8 K) and moderate carrier concentrations (∼1020 cm–3) from charge transport measurements. This work highlights metal flux as a synthetic route to high quality single crystals of novel intermetallic subchalcogenides with Dirac semimetal behavior.
-
Contrasting SnTe–NaSbTe2 and SnTe–NaBiTe2 Thermoelectric Alloys: High Performance Facilitated by Increased Cation Vacancies and Lattice Softening
Journal of the American Chemical Society 2020, 142 (28), 12524-12535. DOI: 10.1021/jacs.0c05650
Abstract
Defect chemistry is critical to designing high performance thermoelectric materials. In SnTe, the naturally large density of cation vacancies results in excessive hole doping and frustrates the ability to control the thermoelectric properties. Yet, recent work also associates the vacancies with suppressed sound velocities and low lattice thermal conductivity, underscoring the need to understand the interplay between alloying, vacancies, and the transport properties of SnTe. Here, we report solid solutions of SnTe with NaSbTe2 and NaBiTe2 (NaSnmSbTem+2 and NaSnmBiTem+2, respectively) and focus on the impact of the ternary alloys on the cation vacancies and thermoelectric properties. We find introduction of NaSbTe2, but not NaBiTe2, into SnTe nearly doubles the natural concentration of Sn vacancies. Furthermore, DFT calculations suggest that both NaSbTe2 and NaBiTe2 facilitate valence band convergence and simultaneously narrow the band gap. These effects improve the power factors but also make the alloys more prone to detrimental bipolar diffusion. Indeed, the performance of NaSnmBiTem+2 is limited by strong bipolar transport and only exhibits modest maximum ZTs ≈ 0.85 at 900 K. In NaSnmSbTem+2 however, the doubled vacancy concentration raises the charge carrier density and suppresses bipolar diffusion, resulting in superior power factors than those of the Bi-containing analogues. Lastly, NaSbTe2 incorporation lowers the sound velocity of SnTe to give glasslike lattice thermal conductivities. Facilitated by the favorable impacts of band convergence, vacancy-augmented hole concentration, and lattice softening, NaSnmSbTem+2 reaches high ZT ≈ 1.2 at 800–900 K and a competitive average ZTavg of 0.7 over 300–873 K. The difference in ZT between two chemically similar compounds underscores the importance of intrinsic defects in engineering high-performance thermoelectrics.

-
Chemical Bonds in Topological Materials
Trends in Chemistry 2021, 3, 9, 700-715. DOI: 10.1016/j.trechm.2021.04.011
Abstract
Topological materials are of great significance in the chemistry, physics, and engineering communities due to their implications for fundamental and applied science. They are solid-state materials where electrons behave atypically, acting as analogs to particles in high-energy physics. In addition, topological materials are impactful in spintronics, catalysis, and quantum information science. However, we do not yet have a holistic understanding of the nature of chemical bonding in these materials, despite growing evidence that it plays a vital role. In this review, we explore how delocalized bonding can lead to topological electronic structures in one-, two-, and three-dimensional systems. We highlight the successes of chemical intuition in polymeric and square-net systems and the potential for one- and three-dimensional structures to follow.

-
Evolving Devil’s Staircase Magnetization from Tunable Charge Density Waves in Nonsymmorphic Dirac Semimetals
Advanced Materials 2021, 33, 41, 2103476. DOI: 10.1002/adma.202103476
Abstract
While several magnetic topological semimetals have been discovered in recent years, their band structures are far from ideal, often obscured by trivial bands at the Fermi energy. Square-net materials with clean, linearly dispersing bands show potential to circumvent this issue. CeSbTe, a square-net material, features multiple magnetic-field-controllable topological phases. Here, it is shown that in this material, even higher degrees of tunability can be achieved by changing the electron count at the square-net motif. Increased electron filling results in structural distortion and formation of charge density waves (CDWs). The modulation wave-vector evolves continuously leading to a region of multiple discrete CDWs and a corresponding complex “Devil’s staircase” magnetic ground state. A series of fractionally quantized magnetization plateaus is observed, which implies direct coupling between CDW and a collective spin-excitation. It is further shown that the CDW creates a robust idealized nonsymmorphic Dirac semimetal, thus providing access to topological systems with rich magnetism.

-
Kinetics and Evolution of Magnetism in Soft-Chemical Synthesis of CrSe2 from KCrSe2
Chemistry of Materials 2021, 33, 20, 8070-8078. DOI: 10.1021/acs.chemmater.1c02620
Abstract
Cation deintercalation with soft-chemical methods provides a route to synthesize new layered compounds with emergent physical and chemical properties that are inaccessible by conventional high-temperature solid-state synthesis methods. One example is CrSe2, a van der Waals (vdW) material that is promising as an air-stable two-dimensional (2D) magnet. Cation deintercalation has rarely been studied mechanistically, and optimized reaction pathways to yield high-quality materials are often poorly understood. In this work, we perform a detailed study of the oxidative deintercalation process of KCrSe2. We prove for the first time using high-resolution scanning transmission electron microscopy (STEM) that even though CrSe2 indeed exists in a true vdW-layered structure, K-intercalated crystalline defects exist in the final product, even when an excess of oxidizing agent was used. We then study the kinetics of the oxidative deintercalation process, showing that it is a zeroth-order reaction with an activation energy of 0.27(6) eV, where the solid-state diffusion of K+ cations in the potassium deintercalation process is the rate-limiting step. Finally, we study the relationship between Cr–Cr distances and the change in magnetic order by tracking how the properties change as a function of varying potassium content due to deintercalation. These data suggest that it might be possible to switch between magnetic states in CrSe2 monolayers by varying its lattice parameters with methods, such as applying strain. Our study also provides a deeper understanding of the cation deintercalation process from a mechanistic perspective that will be helpful for the future development of synthetic methodology that can lead to other new layered materials.

-
A Class of Magnetic Topological Material Candidates with Hypervalent Bi Chains
Journal of the American Chemical Society 2022, 144, 22, 9785-9796. DOI: 10.1021/jacs.2c02281
Abstract
The link between crystal and electronic structure is crucial for understanding structure–property relations in solid-state chemistry. In particular, it has been instrumental in understanding topological materials, where electrons behave differently than they would in conventional solids. Herein, we identify 1D Bi chains as a structural motif of interest for topological materials. We focus on Sm3ZrBi5, a new quasi-one-dimensional (1D) compound in the Ln3MPn5 (Ln = lanthanide; M = metal; Pn = pnictide) family that crystallizes in the P63/mcm space group. Density functional theory calculations indicate a complex, topologically nontrivial electronic structure that changes significantly in the presence of spin–orbit coupling. Magnetic measurements show a quasi-1D antiferromagnetic structure with two magnetic transitions at 11.7 and 10.7 K that are invariant to applied field up to 9 T, indicating magnetically frustrated spins. Heat capacity, electrical, and thermoelectric measurements support this claim and suggest complex scattering behavior in Sm3ZrBi5. This work highlights 1D chains as an unexplored structural motif for identifying topological materials, as well as the potential for rich physical phenomena in the Ln3MPn5 family.

-
Ln3MBi5 (Ln=Pr, Nd, Sm; M=Zr, Hf): Intermetallics with Hypervalent Bismuth Chains
Zeitschrift für anorganische und allgemeine Chemie 2022, 648, 15, e202200123. DOI: 10.1002/zaac.202200123
Abstract
We report five new isostructural compounds in the Ln3MBi5 (Ln=Pr, Nd, Sm; M=Zr, Hf) family, and compare them to the recently reported Sm3ZrBi5 analogue. Ln3MBi5 crystallizes in the P63/mcm space group, hosting the anti-Hf5Sn3Cu structure type. The one-dimensional structure consists of hypervalent Bi2− chains and face-sharing MBi6 octahedra that form chains along the c axis. A framework of Ln3+ cations charge balances and separates the two motifs. The Bi−Bi and M−Bi bond lengths decrease as the Ln cation becomes smaller across the period, but there is almost no difference in either bond length when the identity of M changes, as expected because Zr and Hf have the exact same radii due to lanthanide contraction. We present a structural stability map showing the limits of cation stability in the structure, with La3+ and U3+ as the largest cations and Sm3+ as the smallest cation. X-ray photoelectron spectra suggest ambiguous valence states in the Ln3MBi5 structure depending on the identity of the M atom. This work further expands the Ln3MBi5 family and sheds new light on how their bonding behavior may vary based on chemical composition.

-
Synthesis of an Aqueous, Air-Stable, Superconducting 1T′-WS2 Monolayer Ink
Science Advances 2023, 9, 12, eadd6167. DOI: 10.1126/sciadv.add6167
Abstract
Liquid-phase chemical exfoliation can achieve industry-scale production of two-dimensional (2D) materials for a wide range of applications. However, many 2D materials with potential applications in quantum technologies often fail to leave the laboratory setting because of their air sensitivity and depreciation of physical performance after chemical processing. We report a simple chemical exfoliation method to create a stable, aqueous, surfactant-free, superconducting ink containing phase-pure 1T′-WS2 monolayers that are isostructural to the air-sensitive topological insulator 1T′-WTe2. The printed film is metallic at room temperature and superconducting below 7.3 kelvin, shows strong anisotropic unconventional superconducting behavior with an in-plane and out-of-plane upper critical magnetic field of 30.1 and 5.3 tesla, and is stable at ambient conditions for at least 30 days. Our results show that chemical processing can make nontrivial 2D materials that were formerly only studied in laboratories commercially accessible.
-
Charge Density Wave-Templated Spin Cycloid in Topological Semimetal NdSbxTe2−x−δ
Physical Review Materials 2023, 7, 4, 044203. DOI: 10.1103/PhysRevMaterials.7.044203
Abstract
Magnetic topological semimetals present open questions regarding the interplay of crystal symmetry, magnetism, band topology, and electron correlations. LnSbxTe2−x−δ (Ln denotes Lanthanide) is a family of square-net-derived topological semimetals that allows compositional control of band filling, and access to different topological states via an evolving charge density wave (CDW) distortion. Previously studied Gd and Ce members containing a CDW have shown complex magnetic phase diagrams, which implied that spins localized on Ln interact with the CDW, but to this date no magnetic structures have been solved within the CDW regime of this family of compounds. Here, we report on the interplay of the CDW with magnetism in NdSbxTe2−x−δ by comparing the undistorted square net member NdSb0.94Te0.92 with the CDW-distorted phase NdSb0.48Te1.37, via single-crystal x-ray diffraction, magnetometry, heat capacity, and neutron powder diffraction. NdSb0.94Te0.92 is a collinear antiferromagnet with TN∼2.7K, where spins align antiparallel to each other, but parallel to the square net of the nuclear structure. NdSb0.48Te1.37 exhibits a nearly fivefold-modulated CDW (qCDW=0.18), isostructural to other LnSbxTe2−x−δ at similar x. NdSb0.48Te1.37 displays more complex magnetism with TN=2.3K, additional metamagnetic transitions, and an elliptical cycloid magnetic structure with qmag=−0.41b∗. The magnitudes of qCDW and qmag exhibit an integer relationship, 1+2qmag=qCDW, implying a coupling between the CDW and magnetic structure. Given that the CDW is localized within the nonmagnetic distorted square net, we propose that conduction electrons “template” the spin modulation via the Ruderman-Kittel-Kasuya-Yosida interaction.
-
Acid-Assisted Soft Chemical Route for Preparing High-Quality Superconducting 2M-WS2
Chemistry of Materials 2023, 35, 14, 5487-5496. DOI: 10.1021/acs.chemmater.3c00813
Abstract
2M-WS2 is a metastable, superconducting polymorph of the transition metal dichalcogenide (TMD) WS2, comprised of layers of face-sharing distorted WS6 octahedra. It is predicted to host non-Abelian quantum states, promising for topological computing. Due to its thermodynamic instability, 2M-WS2 cannot be synthesized using solid-state synthesis. Rather, it requires a top-down approach in which K+ is deintercalated from KxWS2; so far, this process has been completed using a strong oxidizer, K2Cr2O7 in dilute H2SO4. A disadvantage of such an indirect synthesis is that the harsh reaction condition may cause the crystal quality to suffer. To date, no studies have been performed to optimize the synthesis or understand the chemical nature of this reaction. In this study, we found that the K-deintercalation process from KxWS2 is spontaneous, and a non-oxidative acidic reaction environment is sufficient to facilitate the oxidation of KxWS2 to 2M-WS2 while reducing H+ to H2. By analyzing the superconducting transition in the heat capacity, we found that 2M-WS2 made using less aggressive methods has higher superconducting volume fractions. We describe how to access the thermodynamically unfavorable superconducting 2M phase of WS2 as high-quality crystals.